
Case Report | DOI: https://doi.org/10.31579/2578-8868/151
1Department of Paediatric Neurology, Children’s Hospital-London Health Sciences Centre, London, ON, CANADA
2Department of Radiology, Children’s Hospital-London Health Sciences Centre, London, ON, CANADA
3Department of Neuroscience, King Faisal Specialist Hospital & Research Center, Riyadh, Saudi Arabia
*Corresponding Author: Sulaiman Almobarak, Department of Paediatric Neurology, Children’s Hospital-London Health Sciences Centre, London, ON, CANADA
Citation: Sulaiman Almobarak, Michael Jurkiewicz, Narayan Prasad. (2021) PelizaeusMerzbacher Disease in a Female due to Proteolipid Protein 1 duplication: A Case Report. J. Neuroscience and Neurological Surgery. 8(1); DOI:10.31579/2578-8868/151
Copyright: © 2021 Sulaiman Almobarak, This is an open-access article distributed under the terms of The Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Received: 23 December 2020 | Accepted: 05 January 2021 | Published: 12 January 2021
Keywords: pelizaeus–merzbacher disease; PMD; PLP1; nystagmus; delayed myelination, hypomyelinating disorders, leukodystrophy.
Pelizaeus–Merzbacher disease (PMD) is a rare X-linked central nervous system disease involving the proteolipid protein 1 (PLP1) gene on Xq22.1. PMD patients’ commonly exhibit signs including nystagmus, hypotonia, and developmental delay. We report a female case of mild spectrum phenotypic expression of PMD attributable to a de novo Copy Number Variant (CNV) change. A two and half-year-old girl presented to our clinic with hypotonicity. She had apneic spells at birth, and was diagnosed to have nystagmus when she was 3 months old. In addition, she presented with delayed motor development including poor head control and inability to sit independently at 6 months of age, eventually standing with support at 20 months, and a prominent wide-based gait at 24 months. MRI head revealed diffuse, markedly delayed myelination, with a reduction in white matter volume. A chromosomal microarray testing indicated that patient carries an Xq22.1 q23 duplication of uncertain significance, of which the PLP1 is fully duplicated. Parental studies were normal. X-inactivation study was normal. Therefore, our case represents a phenotypic expression of PMD due to de novo mutation, a rare occurrence in a female.
Pelizaeus–Merzbacher disease (PMD) is a rare X-linked central nervous system disease involving the proteolipid protein 1 (PLP1) gene on Xq22.1 [1]. VariousPLP1 mutations, including duplications, point mutations, and deletions, lead to oligodendrocyte dysfunction in patients with PMD [2]. PMD is classified according to its severity, with the mild form of PMD the most common, typically presenting with initial symptoms of hypotonia, nystagmus, ataxia and delayed development of motor skills [3,8]. As an X-linked disease, PMD should in theory only affect males [4]. Here we present a female child with PMD due to PLP1 gene duplication and discuss the genetics of disease causation.
The proband is a two and half-year-old girl, the product of a second pregnancy of a non-consanguineous Canadian couple born 40+5 weeks by spontaneous vaginal delivery. There was no family history of other children affected with developmental delay. At birth, she was hypotonic, and developed apneic spells that required resuscitation, and neonatal intensive care unit admission. At 3 to 4 months of age she was diagnosed to have nystagmus. She was also noticed to have delayed motor development: at the age of 5-6 months she had poor head control and was unable to sit independently; at 20 months she was able to crawl, move on her knees, and stand only with support; at 24 months she was able to sit with minimal support and was able to take a few steps with support and a wide-based gait -. She had a vocabulary of few words at that time; at the age of 27 months, she was sitting independently, able to crawl and cruise using a walker. She was poor communicative and remained nonverbal.
On examination, the patient was noted to display axial and appendicular hypotonia, horizontal nystagmus in both eyes, and truncal titubation. There were no clear facial dysmorphic features, but she was noted to have full cheeks and slight bitemporal narrowing. Her head circumference was 45 cm (50thpercentile) weight was 9.4 kg (55th percentile), and height was 78 cm (85th percentile). Deep tendon reflexes were +2 throughout and symmetrical. There were no dystonia or spasticity. There are no feeding difficultiesand no hearing, dental or cardiac concerns. Systems inquiry did not disclose any other concerns.
Her vaccinations were all up-to-date. For the family history, there are no consanguinity between parents or family history of kids with developmental delay.
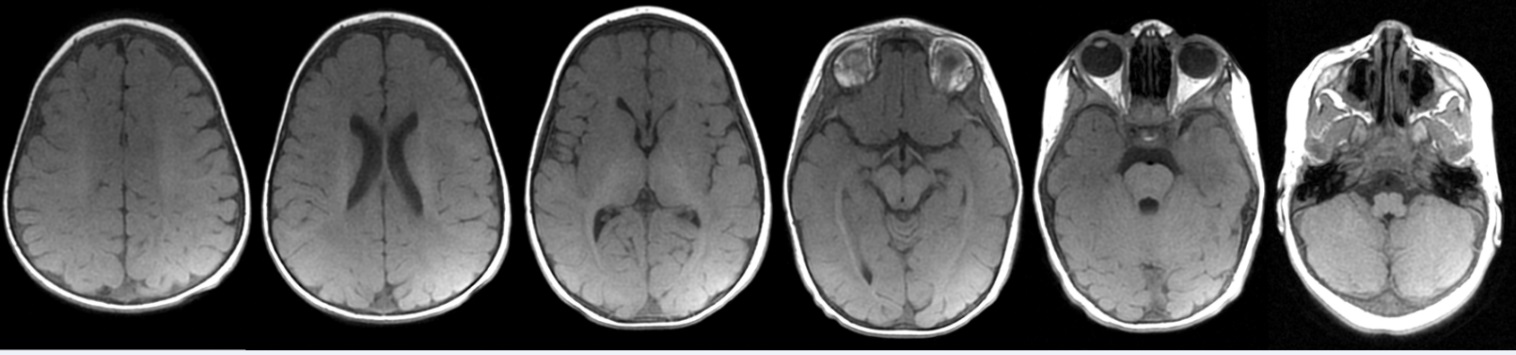
Multiplanar, multi-sequence brain magnetic resonance imaging (MRI) was performed without contrast. T1-weighted images demonstrated abnormal hypointensity in the white matter diffusely, but most prominently in the frontal lobes. T2-weighted images demonstrate abnormal hyperintensity throughout the white matter. At this age, myelination should be complete and the brain MRI should be similar to that of an adult on T1-and T2-weighted images. There was therefore diffuse, markedly delayed myelination, with mildly decreased white matter volume.

Genetic Investigations
A chromosomal microarray testing was performed which disclosed an Xq22.1 q23 duplication of uncertain significance of about 15.4 megabase size involving 73 OMIM genes, of which the PLP1 is fully duplicated. Whole gene duplications of the PLP1 accounts for about 60-70% of patients with PLP1 related disorders, including PMD (OMIM 312080) and spastic-paraplegia type 2 (OMIM 312920) [5]. Individuals with PMD typically have nystagmus, developmental delay, hypotonia, cognitive impairment in addition to leukodystrophy on MRI [3]. The proband in this report has almost all of those features; therefore, the suggestive diagnosis was PMD.
Based on the large size of this duplication and its gene content, this is a pathogenic copy number variant (CNV) in males. Clinical manifestations in female carriers depend on the X inactivation status [8]. Since the X inactivation status was unknown, this CNV was classified as a variant of uncertain significance (VUS). For interpretation, Proband and parental follow-up testing by G-banded chromosome analysis was done to determine the nature of this CNV. X inactivation testing on the proband was also done. The results showed an abnormal female karyotype [46, X, dup(X) (q22.1q23). ish Xq22.3 (RP11-155N17enh) dn. nucish (RP11-155N17x3)] with abnormal molecular cytogenetics. Additional chromosomal material was detected on the long arm of one chromosome X. This additional material is a duplication of bands Xq22.1 through Xq23. All other chromosomes were within normal limits at the level of the resolution achieved. Therefore, the X-inactivation study was considered normal
Fluorescence in Situ Hybridization (FISH) assay was performed using a BAC clone RP11-155N17 which maps to chromosome Xq22.3 nucleotide position chr X: 105459919-105617745 (GRCh37/hg19 assembly). The hybridization pattern was consistent with a tandem duplication at the locus which confirms the duplication first detected by microarray analysis. Parental FISH studies revealed that parental studies were normal, and this copy number variant (CNV) was de novo in origin.
This is a rare case of PMD in a female patient due to duplication of PLP1 gene. PLP1 mutation results in hypomyelination leading to reduced neurological function. PMD is rarely seen in females as it occurs with X-linked recessive inheritance and skewed X inactivation needs to take place [4]. However, our patient developed symptoms associated with PMD in infancy, without X-linked inheritance or skewed X inactivation. Hence, the current genes is depends on one event of gene duplication. Therefore, our case represents a mild spectrum of phenotypic expression of PMD due to de novo mutation.
The current case highlight the importance of performing genetic testing and/or radiological imaging early on for a patient with developmental delay. Our case was diagnosed with PMD at two and half years old due to genetic testing. This early diagnosis was made possible due to patient’s parents who were seeking immediate medical care for their daughter after she showed delayed milestone achievement. In addition to the aforementioned early classic signs and symptoms of PMD, additional PMD signs and symptoms that appear with age are delayed motor skills development, spasticity, stridor, dysarthria, titubation, dystonia, choreiform, and seizures [4, 6]. Though, not all patients will show these signs and symptoms, and they may present as a milder unnoticeable form even if present.
As per the literature, PMD can be diagnosed by MRI as unique features to PMD are distinctively observed on imaging including hypo myelination, increased signal intensity in white matter, corpus callosum thinning, and cerebral hemisphere atrophy [7]. Therefore, for diagnosing PMD, brain MRI is recommended may provide an alternative route to diagnosis with supportive genetic testing for confirmation. Till now no curable treatment is available; treatment is mainly supportive [2]. Some clinical trials of stem cell based treatment and genetics methods are under investigation. [9,10]
We reported a female case affected with PMD due to PLP1 gene duplication which is likewise rare in females. While PMD is best diagnosed by genetic testing, brain MRI may well provide the basis to support its diagnosis in affected individuals with well defined phenotype, where genetic testing may not be accessible.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
